Benefit from 8 unique assets to derisk your solid state!
Insight
Understand why some predicted crystal forms readily crystallize while many others don’t.
JACS 2025
Coverage
Works for neat forms, solvates, salts and co-crystals with 1, 2 and even more molecules per asymmetric unit.
Accuracy
Polymorph stability as a function of Temperature and Relative Humidity with known STandard deviations, TRHu(ST).
Benefit from 8 unique assets to derisk your solid state!
Insight
Understand why some predicted crystal forms readily crystallize while many others don’t.
JACS 2025
Coverage
Works for neat forms, solvates, salts and co-crystals with 1, 2 and even more molecules per asymmetric unit.
Accuracy
Polymorph stability as a function of Temperature and Relative Humidity with known STandard deviations, TRHu(ST).
Expertise
Two decades of experience in finding forms, analyzing failures and improving robustness.
Track record
Best success rates in the last four blind tests on organic crystal structure prediction.
Expertise
Two decades of experience in finding forms, analyzing failures and improving robustness.
Track record
Best success rates in the last four blind tests on organic crystal structure prediction.
On top, did you know that?

ML
Our code also includes Machine Learning well integrated in fully automated work flows. So, our users can focus on their true interest: crystallization science.
Risk
For 30% of the compounds in late development the thermodynamically stable form has not crystallized yet.
Cloud
GRACE runs both on in-house LINUX clusters and in the cloud.
HI (Human Intelligence)
The scientists who write our publications also handle your contract research projects and software support.
There are essentially two complementary ways of accessing
our CSP technology and expertise
Software
Our flagship product GRACE has been developed with and for our customers for over twenty years. From the early days up until today, we have been tightly connected to the experimental departments of some large pharmaceutical companies, embracing their problems as ours.
Our mission is to rush ahead to enable our customers to reliably predict the solid-state properties of their compounds. Since the beginning, we had to run ahead of the crowd: first, competing with numerous academic groups already established in the field of crystal structure prediction and then with other companies, being much larger, benefiting from substantial amounts of venture capital or both. The fact that we never lost our lead definitely proves one thing: we are innovating rapidly and cost-effectively.
We happily integrate third-party components, along with accepting to pay licence fees if that is the best choice for us and our customers. However, for most problems we are facing, we need to go our own way. Our software solutions are robust workflows with a convenient command line interface based on template files. In addition, our workflows run stable for weeks or months on hundreds to thousands of cores, utilizing in-house or cloud High Performance Computing (HPC) Linux clusters. As such, wasting development resources on Graphical User Interfaces (GUIs) or vertical control of all software components is not on our agenda.
Large pharmaceutical companies, who want to apply our technology to their entire small molecule portfolio, often start to use GRACE themselves in-house or on the cloud at some point. GRACE is running on many different LINUX clusters, featuring different types of schedulers (SLURM, SGE, etc). User training has always been straightforward, hardly ever requiring more than 1-2 days of initial training for users who come with some knowledge of organic crystallography and are not afraid of the BASH command line interface. Users can always ask for our help to check the logic and progress of their workflows, but after a few weeks they hardly ever do.
Our current production version, GRACE 3.1, has been used in hundreds of crystal structure prediction studies by internal and external users. We are now preparing to release GRACE 3.2, which will represent yet another major step forward for modern crystal structure prediction.
The commercial release of GRACE currently features the following modules:
Core
The
computing infrastructure.
CSP Factory
A workflow for the fully automated execution
of crystal structure prediction studies and associated tools.
Structure Solution Factory
A workflow for matching large numbers of
predicted crystal structures and their mixtures to experimental powder
diffraction patterns.
Force Field Factory
A workflow, plus tools to generate tailor-made force
fields for third-party applications.
We offer support, maintenance and technical consultancy for all our software products.
We also offer fast-tracked implementation of improvements or new features upon customer request as an additional service.
Interested?
Send a request to quotes@avmatsim.eu
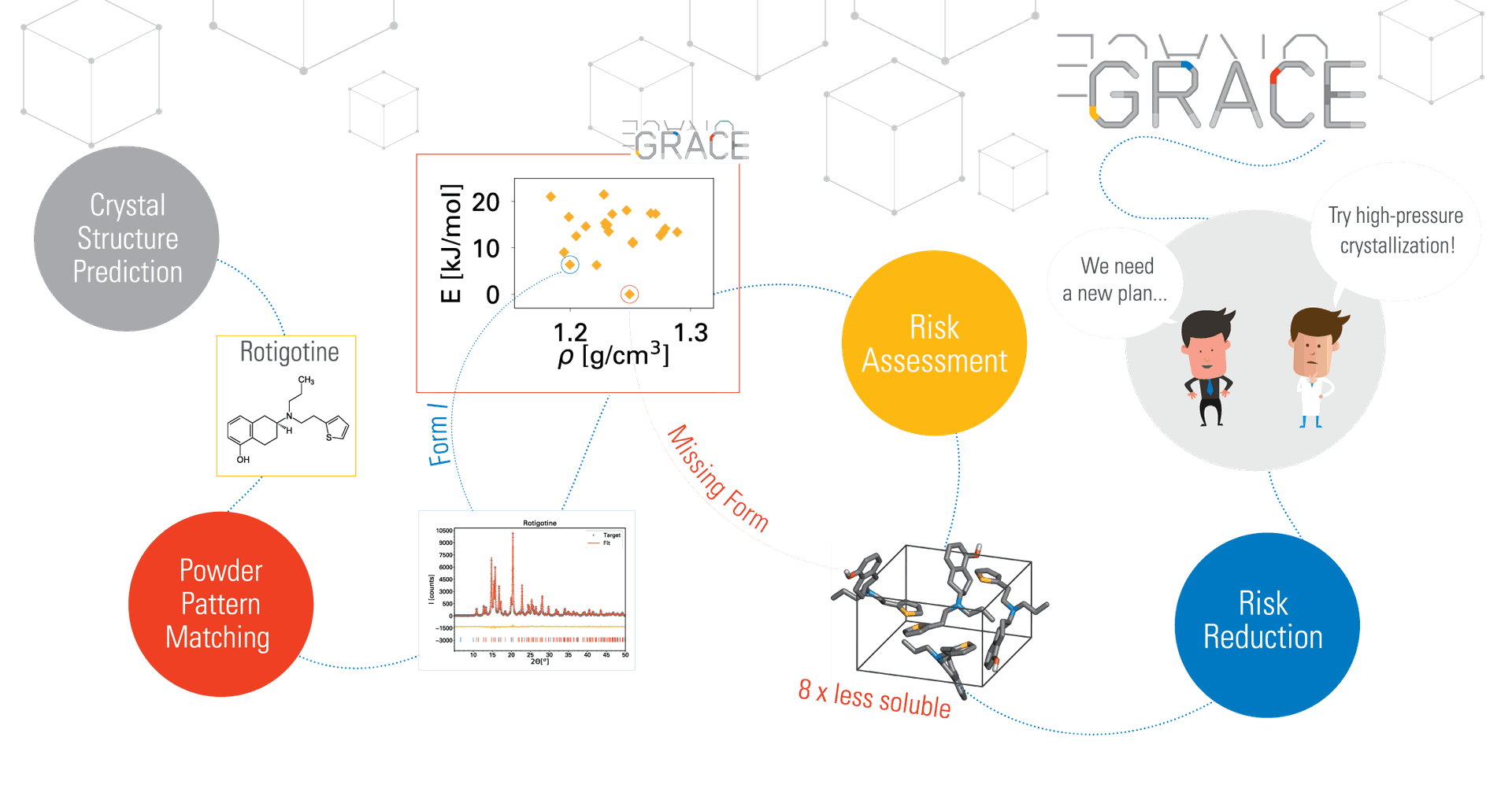
Contract Research
Do you still doubt that...

crystal structures can be
generated from 2D molecular diagrams alone?

crystal structures are easily determined by comparing experimental powder diffraction patterns to predicted structures?

that potentially late-appearing forms can be predicted together with the corresponding solubility change?

or that rational experiment
design can be used to enhance the chances of generating or avoiding such forms?
Our methods work for

neat forms

salts

co-crystals

solvates, and

in particular, hydrates.
Well, our contract research team has been doing all of these things for many years on a daily basis.
Of course, the computational effort grows exponentially with the size, flexibility and number of independent molecules, and so the question is not so much if a project is scientifically feasible, but rather if it is economically reasonable. Because of rapid innovation in hardware and software development, the application domain is redefined every year.
Our computational scientists will not simply dump structures and numbers on you, but will carefully work out what our computational results and your experimental results mean combined with respect to your product development and the risks you are facing.
Our contract research services are the ideal
solution for new customers or those customers having to consider a few
compounds per year. Even customers with a GRACE software license still use our contract
research services in order to benefit from our crystallographic expertise
and
our sharp analysis of solid-state problems, based on experience accumulated through hundreds of crystal structure
prediction projects.
Interested?
Send a request to quotes@avmatsim.eu